

Vererbung beim Charcot-Marie-Tooth-Syndrom
Das CMT-Syndrom wird meist autosomal-dominant, aber auch X-chromosomal oder autosomal-rezessiv vererbt. Seltener kommt es vor, dass die Krankheit das erste Mal innerhalb einer Familie auftritt (= sporadische Fälle). In diesem Fall sind beide Elteren nicht Träger des Krankheitsmerkmals, sondern die massgebende Veränderung im Gen (also die Mutation) ist beim Kind das erste Mal aufgetreten (sozusagen bei der Entstehung des Kindes "passiert"). Für das Kind bedeutet dies aber, dass es seine Erkrankung auf die Nachkommen weitervererben kann.
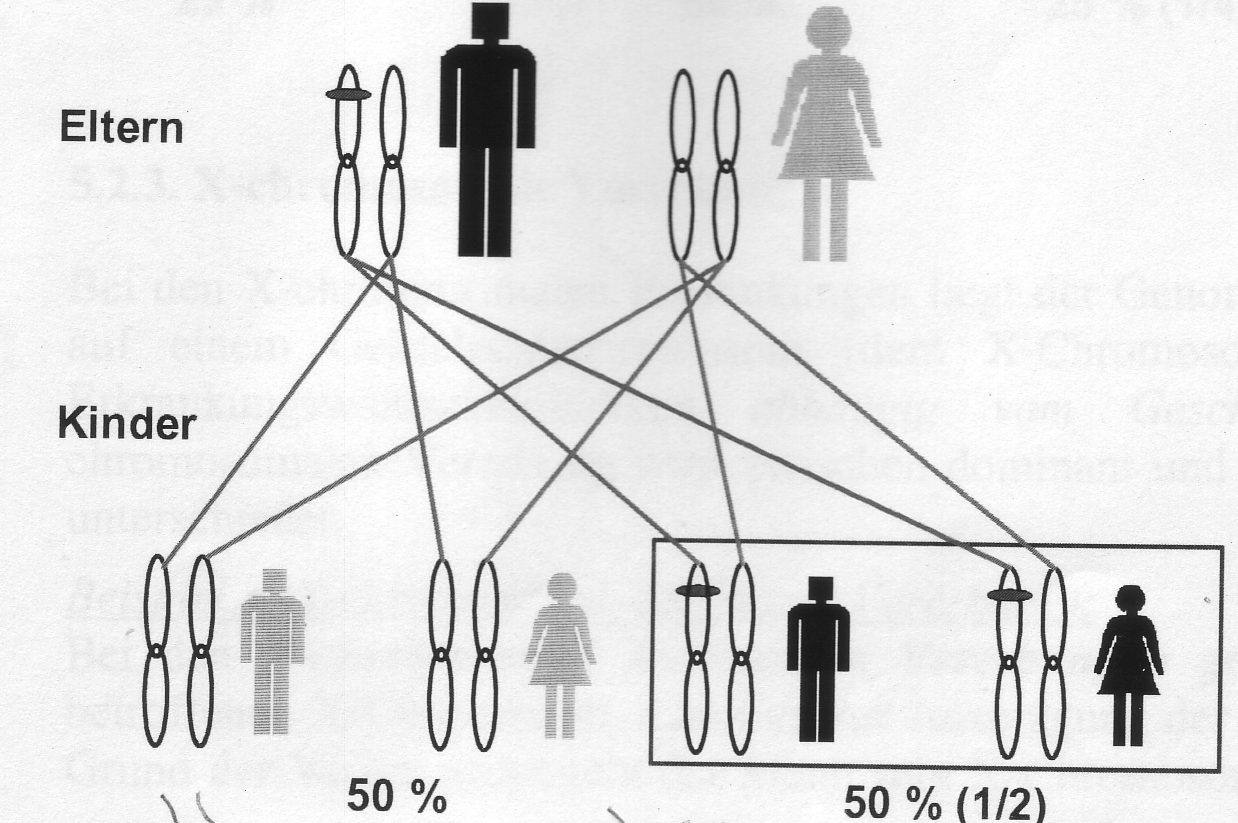
Autosomal-dominante Vererbung
Unter dominanter Vererbung versteht man, dass durch nur eine veränderte Erbanlage (Gen), eine Erkrankung ausbrechen kann. Autosomal-dominant bedeutet, dass die Veränderung auf einem Autosom (Chromosom 1-22) liegt. Ist ein Elternteil erkrankt, besteht eine 50%-ige Wahrscheinlichkeit der Weitervererbung!
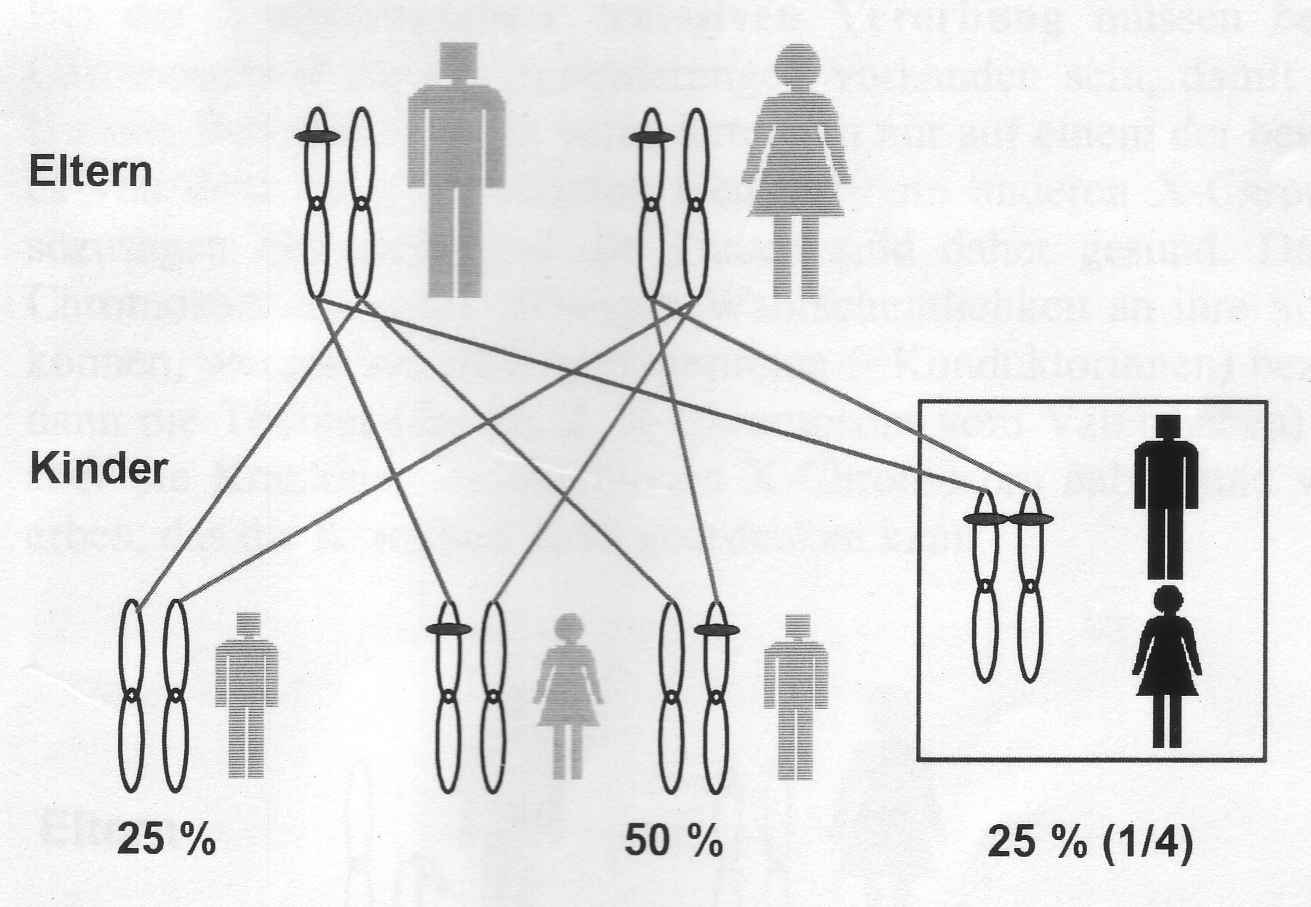
Autosomal-rezessive Vererbung
Autosomal rezessiv bedeutet, dass eine Krankheit nur dann zum Ausbruch kommt, wenn sich auf beiden Chromosomen (1-22) die gleiche Veränderung (Mutation) in einem bestimmten Gen findet, d.h. jeweils eine Veränderung vom Vater und eine von der Mutter geerbt wurde. Die Eltern sind dabei nicht erkrankt.
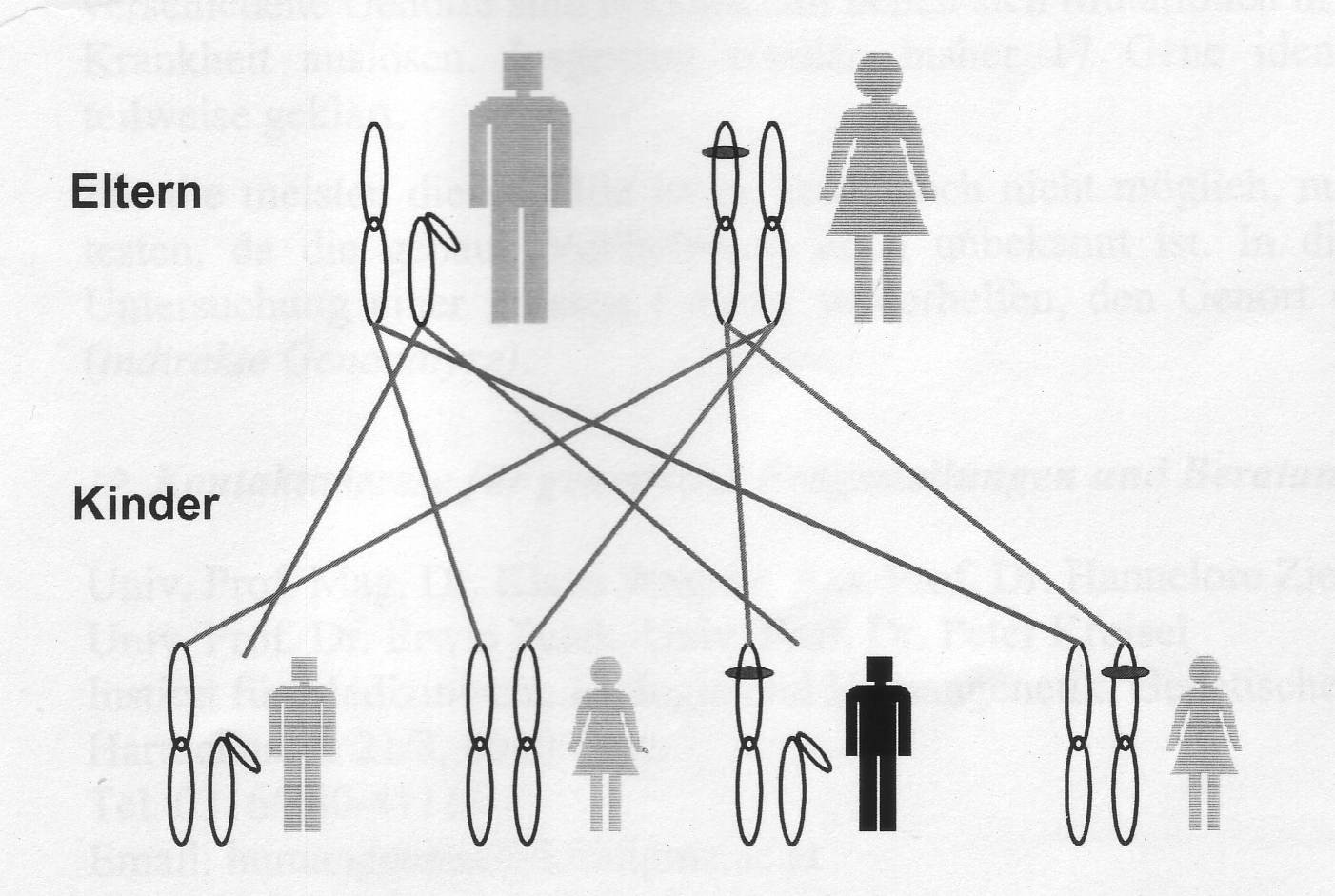
X-chromosomale Vererbungen
Bei den X-chromosomalen Erkrankungen liegt der Genort für die entsprechende Erkrankung auf einem Geschlechtschromosom (dem X-Chromosom) und das bedeutet, dass die Erkrankungswahrscheinlichkeit abhängig vom Geschlecht ist. Auch bei der X-chromosomalen Vererbung wird zwischen dominant und rezessiv wirkenden Veränderungen unterschieden.
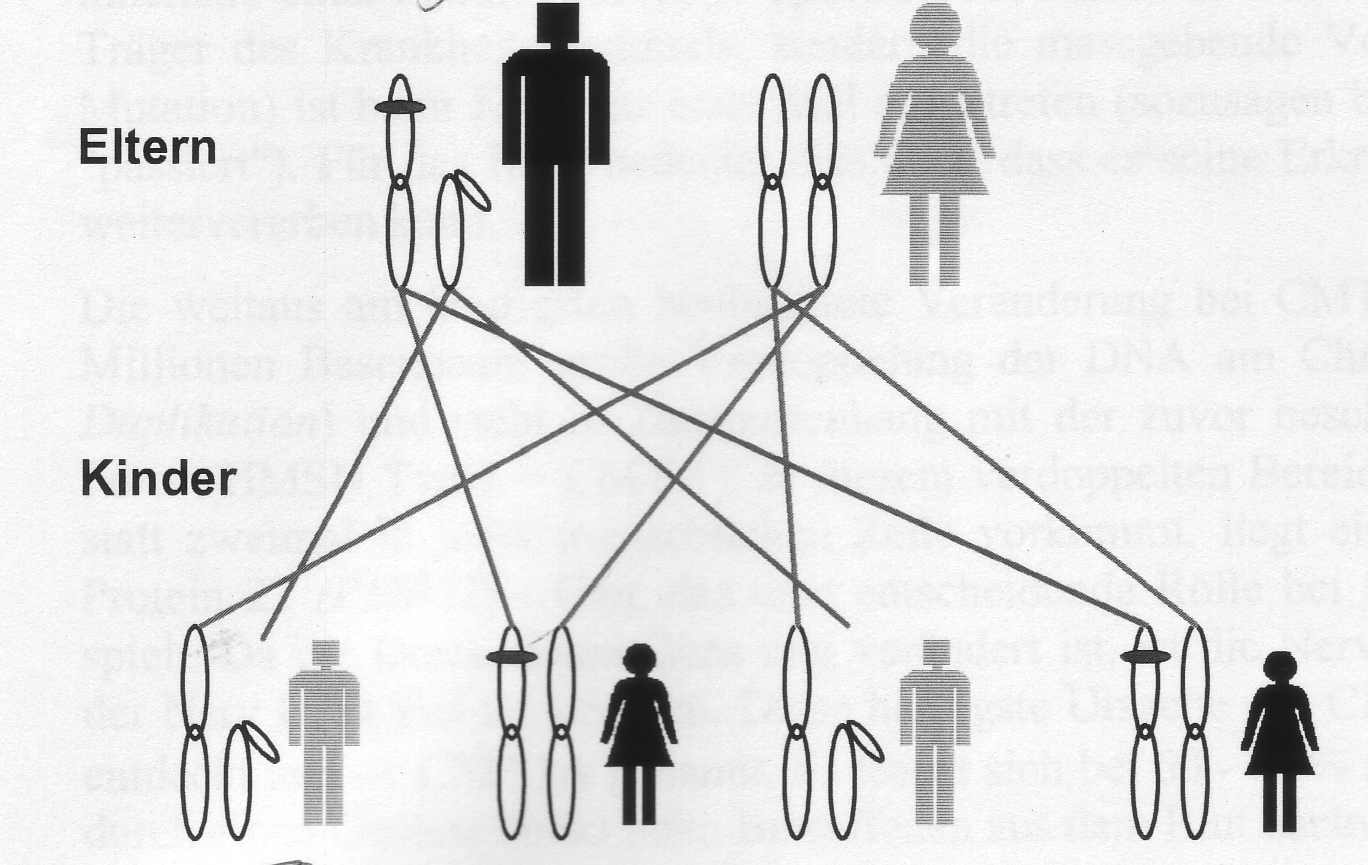
Ist der Vater erkrankt, Weitergabe an die Töchter (defektes X-Chromosom).
Ist die Mutter erkrankt, 50% Wahrscheinlichkeit zur Weitergabe an Söhne und Töchter.
Verdoppelung der DNA am Chromosom 17
Die weitaus am häufigsten beobachtete Veränderung bei CMT-Syndrom ist durch eine 1.5 Millionen Basenpaare große Verdoppelung der DNA am Chromosom 17 bedingt (= sog. Duplikation) Da die Dosis dieses Gens nun verändert ist, ist die Nervenhülle defekt aufgebaut und der Nerv leitet viel zu langsam. Diese häufigste Ursache des CMT-Syndroms wurde als erste entdeckt und ® CMT1A genannt. Es findet sich bei 60 - 70 % aller CMT-Fälle und lässt sich durch einen Gentest direkt beim Betroffenen aus dem Blut nachweisen (direkte Genanalyse).
Fehlendens PMP22-Gen
Am Chromosom 17 kann anstatt der Verdoppelung des PMP22-Gens, dieses auch ganz fehlen und dann also nur auf einem elterlichen Chromosom vorkommen (= sog. Deletion). Dadurch ergibt sich die Sonderform der hereditären Neuropathie mit Neigung zu Druckläsionen (HNPP).
