

Erhebung der Krankengeschichte
Im Gespräch mit dem Arzt ist es wichtig, ihm alle bestehenden Symptome genau anzugeben und hier v.a. auch die zeitliche Reihenfolge des Auftretens zu berichten. Weiters ist es von Bedeutung anzugeben, ob auch bei weiteren Familienmitgliedern Symptome bestehen und in welchem Verwandtschaftsverhältnis diese Personen stehen (= Zeichnen eines Stammbaums)
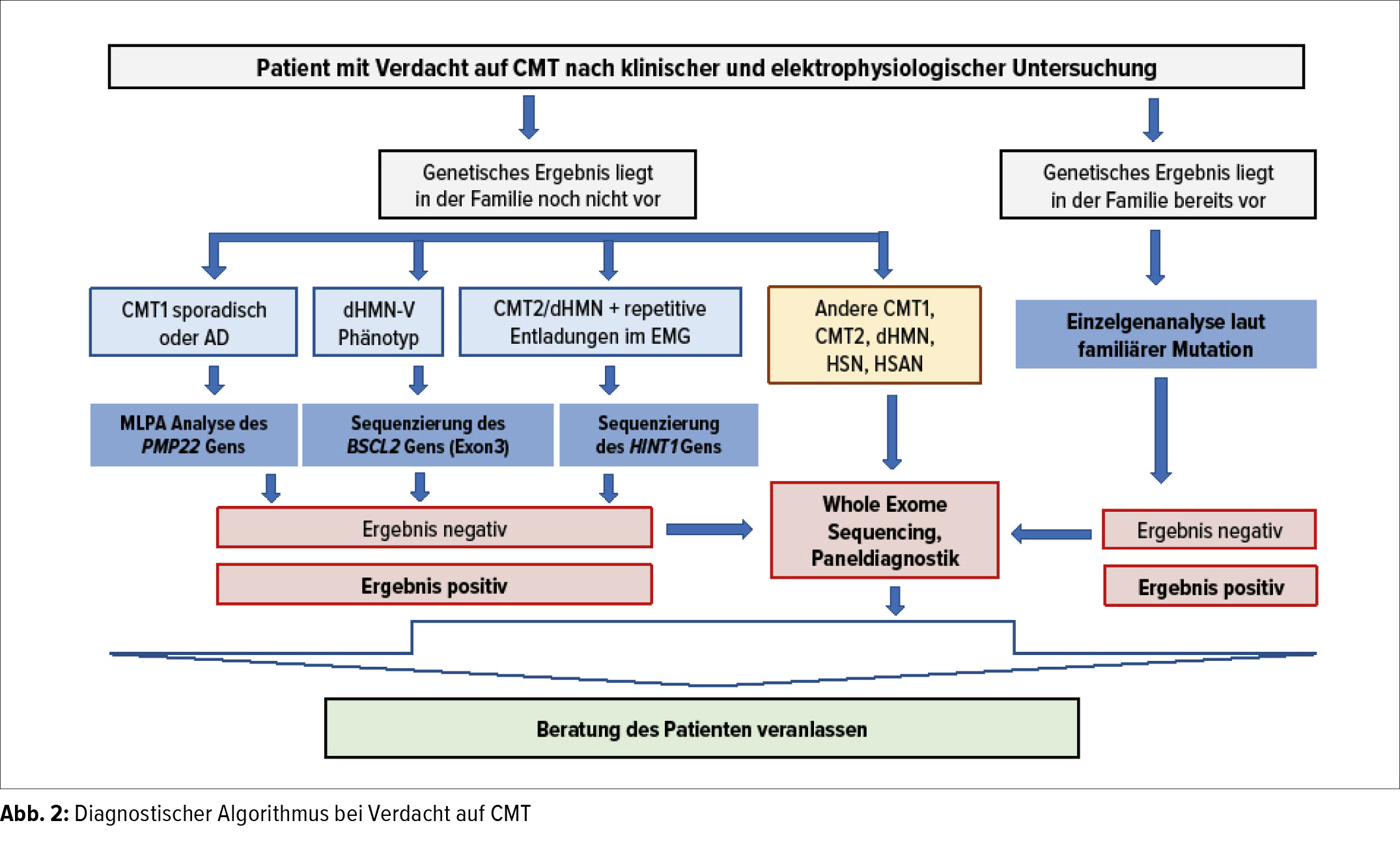
Messung der Nervenleitgeschwindigkeit (NLG)
Hierbei werden Oberflächenelektroden auf einen Muskel aufgeklebt und der Nerv an mehreren Stellen mit Strom gereizt. Meist werden mehrere motorische und sensible Nerven an Händen und Füssen gemessen.
Durch die Bestimmung der Nervenleitgeschwindigkeit (NLG) ist es möglich, das CMT - Syndrom in die 2 wichtigsten (häufigsten) Untergruppen zu unterteilen.
- CMT Typ 1
(NLG < 38 m/sec)
demyelinisierende Form, bedingt durch Veränderungen der Nervenhülle - CMT Typ 2
(NLG > 38 m/sec)
axonale Form, bedingt durch Veränderungen des Nerveninneren (Axons)
(Ein gesunder Nerv erreicht eine NLG von durchschnittlich 50-55 m/sec)
Auch kombinierte Formen (axonal-demyelinisierend) kommen vor, v.a. nach längerem Krankheitsverlauf können beide Anteile des Nervs betroffen sein. Weiters kann durch die Bestimmung der NLG festgelegt werden, ob nur die motorischen oder auch die sensiblen Nervenfasern betroffen sind.
Elektromyographie (EMG)
Die EMG wird mit Nadelektroden durchgeführt, die direkt in den Muskel eingeführt werden. Dies ist manchmal etwas schmerzhaft. Diese Untersuchung ermöglicht einerseits eine Aussage über den Schweregrad der Erkrankung und den aktuellen Verlauf und dient auch in Zweifelsfällen dazu, eine Erkrankung, die direkt durch eine Störung im Muskel verursacht ist (= Myopathie) auszuschliessen.
Magnetevozierte Potentiale (MEP), somatosensible Potentiale (SSEP)
Auch bei diesen beiden Untersuchungen wird elektrisch gereizt und mit Oberflächenelektroden abgeleitet. Sie ermöglichen eine Aussage über die zusätzliche Mitbeteiligung der zentralen motorischen (MEP-Untersuchung) und sensiblen Bahnen (SSEP)
Zuordnung
Wenn diese Untersuchungen abgeschlossen sind, sollte es möglich sein, eine Zuordnung für die weitere genetische Untersuchung zu treffen. Hierbei ist es entscheidend zu differenzieren, ob die Veränderungen:
- die motorischen und sensiblen Nervenfasern betreffen (= hereditäre motorisch-sensible Neuropathie = HMSN, jeweils Typ 1 oder Typ 2 je nach NLG)
- nur oder überwiegend motorische Nervenfasern betreffen (= hereditäre distale motorische Neuropathie = dHMN)
- überwiegend sensible Nervenfasern betroffen sind und eine Neigung zu Wundheilungsstörungen (Ulzera) besteht (= hereditäre sensible-autonome Neuropathie = HSN, HSAN)
- motorische und sensible Nervenfasern betroffen sind und gleichzeitig eine Neigung zu Lähmungen nach Druckexposition oder bereits nach leichter Belastung besteht (= hereditäre Neuropathie mit Neigung zu Druckläsionen = HNPP)
